xTB Nanoreactor
How to prepare a xTB nanoreactor calculation with CREST.
Preparing a xTB nanoreactor calculation
This is the current workaround for the nanoreactor procedure described in JCTC, 2019, 15, 2847-2862.
Currently, there isn’t an automated procedure for the reactor, but the workarounds can be used with the CREST 2.11 version and up. The important functions here are mainly a definition of metadynamics parameters and the logfermi potential used for external compression of the system.
Assuming a given input structure is provided as struc.xyz, there are 3 steps/commands required:
- Generate nano-reactor settings with the command
crest struc.xyz --reactor --genpot <density> --genmtd <sim.length>which will produce a file called
rcontrolcontaining the correctxtbconstraints.<density>can be the required nano-reactor density in g/cm³ like in the JCTC paper,<sim.length>is the metadynamics length in ps. All other settings, for example k and α for the metadynamics, must be directly edited in thercontrolfile. This requires some trial and error but the JCTC paper is generally a good guideline, too. - Run the metadynamics with the generated settings using xtb simply with the command
xtb struc.xyz --gfn 2 --md --input rcontrolThe trajectory is saved as
xtb.trj. - To so some (simple) fragment analyzation of
xtb.trjusecrest struc.xyz --reactor --fragoptThis will extract all fragments from the trajectory based on neighbor lists, optimize their geometry with
xtband sort them.
Example: Benzene dimer
The procedure as described above for the benzene dimer would be as follows.
Assuming we want to perform a nanoreactor MTD simulation with a target density of 7.5 g/cm³ and a simulation length of 10 ps, we get
crest struc.xyz --reactor --genpot 7.5 --genmtd 10
24
C 1.3703175098 -0.2230694787 -0.2051322578
C 0.8724637198 1.0681411469 -0.1985409099
C -0.4946902594 1.2826293364 -0.2020059558
C -1.3639992822 0.2059060662 -0.2120803177
C -0.8661490718 -1.0853142053 -0.2186405367
C 0.5010144102 -1.2998014403 -0.2151588473
H 2.4376123991 -0.3905078209 -0.2004983082
H 1.5510940284 1.9086995897 -0.1890983686
H -0.8833386346 2.2906379550 -0.1953893589
H -2.4313050038 0.3733579606 -0.2130768168
H -1.5448066634 -1.9259014289 -0.2244868410
H 0.8896804372 -2.3078291721 -0.2182027729
C 1.3740605709 -0.2163810201 3.2134083252
C 0.8762063726 1.0748341772 3.2211351784
C -0.4909567838 1.2893253614 3.2169444806
C -1.3602530076 0.2126031859 3.2049588120
C -0.8623947453 -1.0786002367 3.1970726709
C 0.5047555115 -1.2930937341 3.2013278402
H 2.4413668596 -0.3838329632 3.2148252896
H 1.5548574973 1.9154119029 3.2286896265
H -0.8796260526 2.2973473579 3.2212540993
H -2.4275441325 0.3800467544 3.1999735035
H -1.5410146220 -1.9191405817 3.1861705260
H 0.8934089425 -2.3010887124 3.1935809394
==============================================
| |
| C R E S T |
| |
| Conformer-Rotamer Ensemble Sampling Tool |
| based on the GFN methods |
| P.Pracht, S.Grimme |
| Universitaet Bonn, MCTC |
==============================================
Version 2.12, Thu 19. Mai 16:32:32 CEST 2022
Using the xTB program. Compatible with xTB version 6.4.0
Cite work conducted with this code as
• P.Pracht, F.Bohle, S.Grimme, PCCP, 2020, 22, 7169-7192.
• S.Grimme, JCTC, 2019, 15, 2847-2862.
and for works involving QCG as
• S.Spicher, C.Plett, P.Pracht, A.Hansen, S.Grimme,
JCTC, 2022, 18 (5), 3174-3189.
with help from:
C.Bannwarth, F.Bohle, S.Ehlert, S.Grimme,
C.Plett, P.Pracht, S.Spicher
This program is distributed in the hope that it will be useful,
but WITHOUT ANY WARRANTY; without even the implied warranty of
MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE.
Command line input:
> crest struc.xyz --reactor --genpot 7.5 --genmtd 10
Metadynamics settings:
Simulation time: 10.00 ps
Vbias (k): 0.960000 Eh
Vbias (α): 1.000000 Bohr⁻²
Generating spherical logfermi potential:
Reactor density (unscaled): 1.36 g/cm³
Reactor density (from input): 7.50 g/cm³
Spherical cavity radius : 6.157 Bohr
Logfermi temperature : 6000.0 K
Base settings written to file <rcontrol>
$md
time=10.00
step=1.0
shake=0
$set
mddump 2000
$metadyn
save=10
kpush=0.960000
alp=1.000000
$wall
potential=logfermi
sphere: 6.157153582516958, all
temp=6000.0
$end
As can be seen in the rcontrol tab above, the metadynamics parameters k and α have been assigned just some default values of 0.96 Eh and 1.0 Bohr-2, respectively. The target density of 7.5 cm/g³ was automatically converted into the sphere cavity of the $wall potential. All settings can (and should) be adjusted by the user by editing the rcontrol file depending on their needs and the investigated system. As mentioned previously, this is an trial and error process.
With the prepared rcontrol file the xtb metadynamics calculation at GFN2-xTB level is started via
xtb struc.xyz --gfn 2 --md --input rcontrol
and provides the xtb.trj trajectory file.
xtb metadynamics calculation crashes, parameters in the rcontrol file must be adjusted!If desired, the trajectory snapshots can be optimized and sorted via CREST. This is an optional refinement step. Each snapshot is analyzed with regards to its molecular topology. If any change/fragmentation is detected, the respective snapshot is selected for reoptimization. A summary depending on the molecular sum formula is printed at the end, and all products are written to an ensemble file called crest_products.xyz Note, that this ensemble file does not satisfy CREST Input Format conventions since molecules with different composition/numbers of atoms are included there.
crest struc.xyz --reactor --fragopt
==============================================
| |
| C R E S T |
| |
| Conformer-Rotamer Ensemble Sampling Tool |
| based on the GFN methods |
| P.Pracht, S.Grimme |
| Universitaet Bonn, MCTC |
==============================================
Version 2.12, Thu 19. Mai 16:32:32 CEST 2022
Using the xTB program. Compatible with xTB version 6.4.0
Cite work conducted with this code as
• P.Pracht, F.Bohle, S.Grimme, PCCP, 2020, 22, 7169-7192.
• S.Grimme, JCTC, 2019, 15, 2847-2862.
and for works involving QCG as
• S.Spicher, C.Plett, P.Pracht, A.Hansen, S.Grimme,
JCTC, 2022, 18 (5), 3174-3189.
with help from:
C.Bannwarth, F.Bohle, S.Ehlert, S.Grimme,
C.Plett, P.Pracht, S.Spicher
This program is distributed in the hope that it will be useful,
but WITHOUT ANY WARRANTY; without even the implied warranty of
MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE.
Command line input:
> crest struc.xyz --reactor --fragopt
========================================
| GFNn-xTB NANOREACTOR |
| SG, Universitaet Bonn, MCTC |
========================================
JCTC, 2019, 15, 2847-2862.
Trajectory file: xtb.trj
Number of atoms = 24
Number of snapshots = 200
<Comparing structures>
Taken = T T T T T T T T T T T T T T T T T T T T T T T T T T T T T T F T T T F
F F F F T T F F T T F T T T F T T T F F F F T T T F T T T T T F T F F T T T F F
F F F F T T T T T T T T T F T T T T T F F F F F F T T T F T T F T T T T F T T T
T T F F F T T T T T T T T T T T T F T F T F T T T T T T T F T F T T T T T T T T
T T T T T F F T T T T T T T T T T T T F T T T T T T T T T T F F T T T T T T T T
T T T T T
153 of 200 taken.
-------------------------
optimization of fragments
-------------------------
Starting optimization of reactor products
155 jobs to do.
[....]
done.
------------------------
reactor products summary
------------------------
structure #atoms Etot composition
1 12 -15.87963968 H6C6
2 13 -16.33341423 H7C6
3 8 -13.60339736 H2C6
4 24 -31.65866501 H12C12
5 24 -31.63609714 H12C12
6 24 -31.61072033 H12C12
7 24 -31.61027908 H12C12
8 24 -31.75015232 H12C12
9 23 -31.21831720 H11C12
10 23 -31.20327344 H11C12
11 24 -31.74950806 H12C12
12 24 -31.66455867 H12C12
13 24 -31.66626426 H12C12
14 24 -31.76250970 H12C12
Structures written to file "crest_products.xyz"
-----------------
Wall Time Summary
-----------------
nano reactor wall time : 0h : 0m :11s
--------------------
Overall wall time : 0h : 0m :11s
CREST terminated normally.
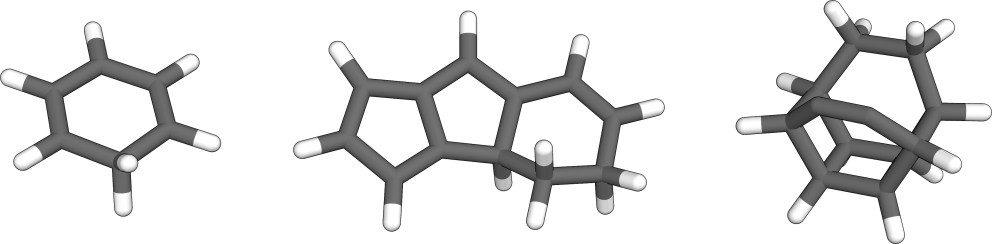
Several interesting nanoreactor products were found for the benzene dimer, starting from simple proton transfers, up to the formation of polycyclic molecules.