QCG Example 2
An example for generating an ensemble of microsolvation structures.
Generating an ensemble of microsolvation structures
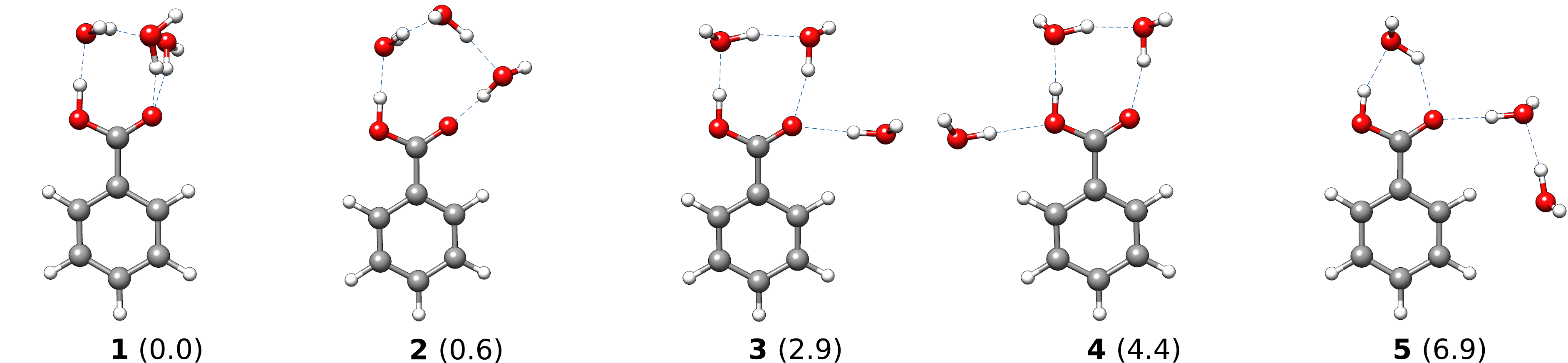
Creating ensembles of generated clusters is important for various reasons. For example, the conformational space is explored during the used MD and MTD simulations so that new energy minima are usually found. Additionally, many problems require the weighting of different populated structures and the inclusion of the conformational entropy. As an example, a microsolvation approach is considered, but also large ensembles with multiple solvent shells can be generated similarly. As typically only a few solvents are added for this, the conformational space is rather small and it is possible to find relatively complete ensembles within a reasonable computational time. Now we want to add three water molecules to benzoic acid. For this, we again provide solute as well as solvent coordinates and call for the ensemble generation.
crest benzoic_acid.xyz --qcg water.xyz --nsolv 3 --T 12 --ensemble --mdtime 50 --alpb water --wscal 1.0 --nofix
15
H -5.151895 0.608937 0.184841
C -4.075803 0.560948 0.103703
C -3.304923 1.648961 0.482499
H -3.781062 2.542533 0.858155
C -1.927760 1.593624 0.380574
H -1.316613 2.433539 0.671921
C -1.315885 0.440886 -0.104813
C 0.159025 0.350784 -0.229059
O 0.718993 -0.633914 -0.685096
O 0.806733 1.411370 0.189344
C -2.093917 -0.650077 -0.484577
H -1.601704 -1.534740 -0.859582
C -3.469918 -0.588324 -0.379395
H -4.072688 -1.434587 -0.673879
H 1.807623 1.318950 0.057503
3
O -0.1918040235 1.3862489483 0.0047370042
H 0.7660977787 1.3911615443 -0.0141642652
H -0.4927337474 1.6150799341 -0.8756928250
==============================================
| |
| C R E S T |
| |
| Conformer-Rotamer Ensemble Sampling Tool |
| based on the GFN methods |
| P.Pracht, S.Grimme |
| Universitaet Bonn, MCTC |
==============================================
Version 2.11.1, Mon 16. Aug 09:59:32 CEST 2021
Using the xTB program. Compatible with xTB version 6.4.0
Cite work conducted with this code as
P. Pracht, F. Bohle, S. Grimme, PCCP, 2020, 22, 7169-7192.
and S. Grimme, JCTC, 2019, 15, 2847-2862.
with help from:
C.Bannwarth, F.Bohle, S.Ehlert, S.Grimme,
C. Plett, P.Pracht, S. Spicher
This program is distributed in the hope that it will be useful,
but WITHOUT ANY WARRANTY; without even the implied warranty of
MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE.
Command line input:
> crest benzoic_acid.xyz --qcg water.coord --nsolv 3 -T 12 -ensemble -mdtime 50 --alpb water --wscal 1.0 --nofix
Solute-file: benzoic_acid.xyz
Solvent-file: water.coord
-T 12 (CPUs/Threads selected)
-mdtime 50 (MD length in ps)
--alpb water : implicit solvation
========================================
| ---------------- |
| Q C G |
| ---------------- |
| Quantum Cluster Growth |
| University of Bonn, MCTC |
========================================
S. Grimme, S. Spicher, unpublished.
=========================================
| quantum cluster growth: INPUT |
=========================================
QCG: Cluster + Ensemble Generation
Ensemble generated via CREST
input parameters
solute : benzoic_acid.xyz
charge : 0
uhf : 0
solvent : water.coord
# of solvents to add : 3
Cluster generated that are above 10 % populated
# of CPUs used : 12
Solvation model : water
xtb opt level : normal
System temperature [K] : 298.0
RRHO scaling factor : 0.75
Solute geometry
molecular radius (Bohr**1): 6.57
molecular area (Bohr**2): 635.98
molecular volume (Bohr**3): 1188.36
Solvent geometry
molecular radius (Bohr**1): 3.88
molecular area (Bohr**2): 194.90
molecular volume (Bohr**3): 244.27
radius of solute : 10.59
radius of solvent : 6.25
=========================================
| Preoptimization |
=========================================
-------------------------
xTB Geometry Optimization
-------------------------
Geometry successfully optimized.
Generating LMOs for solute
Total Energy of solute: -26.1730317 Eh
-------------------------
xTB Geometry Optimization
-------------------------
Geometry successfully optimized.
Generating LMOs for solvent
Total energy of solvent: -5.0705444 Eh
________________________________________________________________________
__________________ Solute Cluster Generation _____________________
________________________________________________________________________
=========================================
| quantum cluster growth: GROW |
=========================================
Solute:
unit ellipsoid axis a,b,c : 0.408 0.306 0.286
Solvent:
unit ellipsoid axis a,b,c : 0.386 0.322 0.292
solvent anisotropy : 1.133
solute anisotropy : 1.169
roff inner wall : 1.388
solute max dist : 17.497
solvent max dist : 7.283
inner unit axis : 0.487 0.274 0.239
inner ellipsoid/Bohr: 14.890 8.363 7.292
outer ellipsoid/Bohr: 14.686 11.006 10.277
Size E /Eh De/kcal Detot/kcal Density Efix R av/act. Surface Opt
1 -31.277550 -21.32 -21.32 1.155 -7.372 0.0 0.0 1359.9 normal
2 -36.366081 -11.29 -32.61 1.143 -7.936 9.0 7.9 1551.0 normal
3 -41.458471 -13.71 -46.31 1.148 -8.463 9.1 10.0 1720.2 normal
Growth finished after 3 solvents added
Results can be found in grow directory
Energy list on file 'qcg_energy.dat'
Interaction energy on file 'qcg_conv.dat'
Growing process on 'qcg_grow.xyz'
Final geometry after grow in 'cluster.coord' and 'cluster.xyz'
Potentials and geometry written in 'cluster_cavity.coord' and 'twopot_cavity.coord'
=========================================
| quantum cluster growth: ENSEMBLE |
=========================================
Method for ensemble search:--gff
Starting ensemble cluster generation by CREST routine
------------------------------------------------
Generating MTD length from a flexibility measure
------------------------------------------------
System flexiblity is set to 1.0 for NCI mode
flexibility measure : 1.000
t(MTD) / ps set by command line : 50.0
t(MTD) / ps : 50.0
Σ(t(MTD)) / ps : 600.0 (12 MTDs)
-------------------------------------
Starting a trial MTD to test settings
-------------------------------------
Estimated runtime for one MTD (50.0 ps) on a single thread: 1 min 15 sec
Estimated runtime for a batch of 12 MTDs on 12 threads: 1 min 15 sec
list of Vbias parameters applied:
$metadyn 0.00125 1.000
$metadyn 0.00083 1.000
$metadyn 0.00056 1.000
$metadyn 0.00125 0.667
$metadyn 0.00083 0.667
$metadyn 0.00056 0.667
$metadyn 0.00125 0.444
$metadyn 0.00083 0.444
$metadyn 0.00056 0.444
$metadyn 0.00125 0.296
$metadyn 0.00083 0.296
$metadyn 0.00056 0.296
*******************************************************************************************
** N E W I T E R A T I O N C Y C L E **
*******************************************************************************************
========================================
MTD Iteration 1
========================================
========================================
| Meta-MD (MTD) Sampling |
========================================
Starting Meta-MD 1 with the settings:
MD time /ps : 50.0
dt /fs : 1.5
dumpstep(trj) /fs : 200
dumpstep(Vbias)/ps : 1.0
Vbias factor k /Eh : 0.0300
Vbias exp α /bohr⁻²: 1.00
Starting Meta-MD 2 with the settings:
MD time /ps : 50.0
dt /fs : 1.5
dumpstep(trj) /fs : 200
dumpstep(Vbias)/ps : 1.0
Vbias factor k /Eh : 0.0200
Vbias exp α /bohr⁻²: 1.00
Starting Meta-MD 3 with the settings:
MD time /ps : 50.0
dt /fs : 1.5
dumpstep(trj) /fs : 200
dumpstep(Vbias)/ps : 1.0
Vbias factor k /Eh : 0.0133
Vbias exp α /bohr⁻²: 1.00
Starting Meta-MD 4 with the settings:
MD time /ps : 50.0
dt /fs : 1.5
dumpstep(trj) /fs : 200
dumpstep(Vbias)/ps : 1.0
Vbias factor k /Eh : 0.0300
Vbias exp α /bohr⁻²: 0.67
Starting Meta-MD 5 with the settings:
MD time /ps : 50.0
dt /fs : 1.5
dumpstep(trj) /fs : 200
dumpstep(Vbias)/ps : 1.0
Vbias factor k /Eh : 0.0200
Vbias exp α /bohr⁻²: 0.67
Starting Meta-MD 6 with the settings:
MD time /ps : 50.0
dt /fs : 1.5
dumpstep(trj) /fs : 200
dumpstep(Vbias)/ps : 1.0
Vbias factor k /Eh : 0.0133
Vbias exp α /bohr⁻²: 0.67
Starting Meta-MD 7 with the settings:
MD time /ps : 50.0
dt /fs : 1.5
dumpstep(trj) /fs : 200
dumpstep(Vbias)/ps : 1.0
Vbias factor k /Eh : 0.0300
Vbias exp α /bohr⁻²: 0.44
Starting Meta-MD 8 with the settings:
MD time /ps : 50.0
dt /fs : 1.5
dumpstep(trj) /fs : 200
dumpstep(Vbias)/ps : 1.0
Vbias factor k /Eh : 0.0200
Vbias exp α /bohr⁻²: 0.44
Starting Meta-MD 9 with the settings:
MD time /ps : 50.0
dt /fs : 1.5
dumpstep(trj) /fs : 200
dumpstep(Vbias)/ps : 1.0
Vbias factor k /Eh : 0.0133
Vbias exp α /bohr⁻²: 0.44
Starting Meta-MD 12 with the settings:
MD time /ps : 50.0
dt /fs : 1.5
dumpstep(trj) /fs : 200
dumpstep(Vbias)/ps : 1.0
Vbias factor k /Eh : 0.0133
Vbias exp α /bohr⁻²: 0.30
Starting Meta-MD 11 with the settings:
MD time /ps : 50.0
dt /fs : 1.5
dumpstep(trj) /fs : 200
dumpstep(Vbias)/ps : 1.0
Vbias factor k /Eh : 0.0200
Vbias exp α /bohr⁻²: 0.30
Starting Meta-MD 10 with the settings:
MD time /ps : 50.0
dt /fs : 1.5
dumpstep(trj) /fs : 200
dumpstep(Vbias)/ps : 1.0
Vbias factor k /Eh : 0.0300
Vbias exp α /bohr⁻²: 0.30
*Meta-MTD 8 finished*
*Meta-MTD 3 finished*
*Meta-MTD 5 finished*
*Meta-MTD 6 finished*
*Meta-MTD 4 finished*
*Meta-MTD 2 finished*
*Meta-MTD 1 finished*
*Meta-MTD 10 finished*
*Meta-MTD 12 finished*
*Meta-MTD 9 finished*
*Meta-MTD 11 finished*
*Meta-MTD 7 finished*
-----------------------
Multilevel Optimization
-----------------------
-------------------------
1. crude pre-optimization
-------------------------
Optimizing all 3000 structures from file "crest_rotamers_0.xyz" ...
1 [...] 3000
done.
input file name : crest_rotamers_1.xyz
output file name : crest_rotamers_2.xyz
reference state Etot : -4.01804455000000
3000 structures remain within 6.00 kcal/mol window
========================================
MTD Iteration 2
========================================
========================================
| Meta-MD (MTD) Sampling |
========================================
Starting Meta-MD 1 with the settings:
MD time /ps : 50.0
dt /fs : 1.5
dumpstep(trj) /fs : 200
dumpstep(Vbias)/ps : 1.0
Vbias factor k /Eh : 0.0300
Vbias exp α /bohr⁻²: 1.00
[...]
Starting Meta-MD 9 with the settings:
MD time /ps : 50.0
dt /fs : 1.5
dumpstep(trj) /fs : 200
dumpstep(Vbias)/ps : 1.0
Vbias factor k /Eh : 0.0133
Vbias exp α /bohr⁻²: 0.44
*Meta-MTD 3 finished*
*Meta-MTD 9 finished*
*Meta-MTD 7 finished*
*Meta-MTD 5 finished*
*Meta-MTD 1 finished*
*Meta-MTD 2 finished*
*Meta-MTD 6 finished*
*Meta-MTD 10 finished*
*Meta-MTD 4 finished*
*Meta-MTD 8 finished*
-----------------------
Multilevel Optimization
-----------------------
-------------------------
1. crude pre-optimization
-------------------------
Optimizing all 2500 structures from file "crest_rotamers_0.xyz" ...
1 [...] 2500
done.
input file name : crest_rotamers_1.xyz
output file name : crest_rotamers_2.xyz
reference state Etot : -4.01784771000000
2500 structures remain within 6.00 kcal/mol window
========================================
MTD Iterations done
========================================
Collecting ensmbles.
running RMSDs...
done.
E lowest : -4.01804
142 structures remain within 3.00 kcal/mol window
-----------------------------------------------
Additional regular MDs on lowest 3 conformer(s)
-----------------------------------------------
Starting MD 1 with the settings:
MD time /ps : 25.0
MD Temperature /K : 400.0
dt /fs : 1.5
dumpstep(trj) /fs : 200
[...]
Starting MD 6 with the settings:
MD time /ps : 25.0
MD Temperature /K : 500.0
dt /fs : 1.5
dumpstep(trj) /fs : 200
*MD 4 finished*
*MD 1 finished*
*MD 2 finished*
*MD 6 finished*
*MD 3 finished*
*MD 5 finished*
Appending file crest_rotamers_1.xyz with new structures
--------------------------------------------
Ensemble optimization with normal thresholds
--------------------------------------------
Optimizing all 892 structures from file "crest_rotamers_1.xyz" ...
1 [...] 892
done.
input file name : crest_rotamers_2.xyz
output file name : crest_rotamers_3.xyz
reference state Etot : -4.01951159000000
892 structures remain within 3.00 kcal/mol window
...............................................
A new lower conformer was found!
Improved by 0.00147 Eh or 0.92058kcal/mol
...............................................
*******************************************************************************************
** N E W I T E R A T I O N C Y C L E **
*******************************************************************************************
========================================
MTD Iteration 1
========================================
========================================
| Meta-MD (MTD) Sampling |
========================================
Starting Meta-MD 1 with the settings:
MD time /ps : 50.0
dt /fs : 1.5
dumpstep(trj) /fs : 200
dumpstep(Vbias)/ps : 1.0
Vbias factor k /Eh : 0.0300
Vbias exp α /bohr⁻²: 1.00
[...]
Starting Meta-MD 9 with the settings:
MD time /ps : 50.0
dt /fs : 1.5
dumpstep(trj) /fs : 200
dumpstep(Vbias)/ps : 1.0
Vbias factor k /Eh : 0.0133
Vbias exp α /bohr⁻²: 0.44
*Meta-MTD 3 finished*
*Meta-MTD 7 finished*
*Meta-MTD 5 finished*
*Meta-MTD 4 finished*
*Meta-MTD 1 finished*
*Meta-MTD 8 finished*
*Meta-MTD 2 finished*
*Meta-MTD 6 finished*
*Meta-MTD 9 finished*
*Meta-MTD 10 finished*
-----------------------
Multilevel Optimization
-----------------------
-------------------------
1. crude pre-optimization
-------------------------
Optimizing all 2500 structures from file "crest_rotamers_0.xyz" ...
1 [...] 2500
done.
input file name : crest_rotamers_1.xyz
output file name : crest_rotamers_2.xyz
reference state Etot : -4.01859099000000
2500 structures remain within 6.00 kcal/mol window
========================================
MTD Iterations done
========================================
Collecting ensmbles.
running RMSDs...
done.
E lowest : -4.01859
77 structures remain within 3.00 kcal/mol window
-----------------------------------------------
Additional regular MDs on lowest 3 conformer(s)
-----------------------------------------------
Starting MD 1 with the settings:
MD time /ps : 25.0
MD Temperature /K : 400.0
dt /fs : 1.5
dumpstep(trj) /fs : 200
[...]
Starting MD 6 with the settings:
MD time /ps : 25.0
MD Temperature /K : 500.0
dt /fs : 1.5
dumpstep(trj) /fs : 200
*MD 5 finished*
*MD 6 finished*
*MD 4 finished*
*MD 2 finished*
*MD 1 finished*
*MD 3 finished*
Appending file crest_rotamers_1.xyz with new structures
--------------------------------------------
Ensemble optimization with normal thresholds
--------------------------------------------
Optimizing all 827 structures from file "crest_rotamers_1.xyz" ...
1 [...] 827
done.
input file name : crest_rotamers_2.xyz
output file name : crest_rotamers_3.xyz
reference state Etot : -4.01950240000000
827 structures remain within 3.00 kcal/mol window
================================================
| Final Geometry Optimization |
================================================
--------------------------------------------
Ensemble optimization with normal thresholds
--------------------------------------------
Optimizing all 827 structures from file "crest_rotamers_3.xyz" ...
1 [...] 827
done.
input file name : crest_rotamers_4.xyz
output file name : crest_rotamers_5.xyz
reference state Etot : -4.01950637000000
827 structures remain within 3.00 kcal/mol window
GFN2-xTB optimization
--------------------------------------------
Ensemble optimization with normal thresholds
--------------------------------------------
Optimizing all 827 structures from file "crest_rotamers_5.xyz" ...
1 [..] 827
done.
-------------------------------------------
Ensemble optimization with tight thresholds
-------------------------------------------
Optimizing all 827 structures from file "crest_rotamers_6.xyz" ...
1 [...] 827
done.
Single point computation with GBSA model
827 jobs to do.
done.
Cluster E /Eh Density Efix R av/act. Surface Opt
1 -41.458562 1.139 0.000 9.6 8.3 935.1 tight
[...]
827 -41.446970 1.121 0.000 6.5 7.9 926.7 tight
Conformers taken: 10
------------------------------------------------------------------------
------------------------------------------------------------------------
Boltz. averaged energy of final cluster:
G /Eh : -41.46409513
T*S /kcal : -1.364
Ensemble generation finished.
Results can be found in ensemble directory
Lowest energy conformer on file 'crest_best.xyz'
List of full ensemble on file 'full_ensemble.xyz'
List of used ensemble on file 'final_ensemble.xyz'
Thermodynamical data on file 'thermo_data'
Population of full ensemble on file 'full_population.dat'
Population on file 'population.dat'
-----------------
Wall Time Summary
-----------------
test MD wall time : 0h : 0m : 0s
MTD wall time : 0h : 0m :40s
multilevel OPT wall time : 0h : 2m :23s
MD wall time : 0h : 5m :56s
--------------------
Overall wall time : 0h : 9m : 8s
CREST terminated normally.
To make sure that we have a reasonable ensemble and energy minima, the MTD time was set to 50 ps. The ALPB solvent model was used to have a better energy ranking of the ensemble structures. It is only applied during final single-point computations. As the solvent is water, we used the –nofix flag so that the solute is not fixed during the Growth. Also, the scaling factor for the outer wall potential was set to 1.0.
The result of the above procedure will be
- an ensemble, written to
full_ensemble.xyz - the energetically lowest structure to
crest_best.xyz - and a file constaining populations of the clusters
full_population.dat.