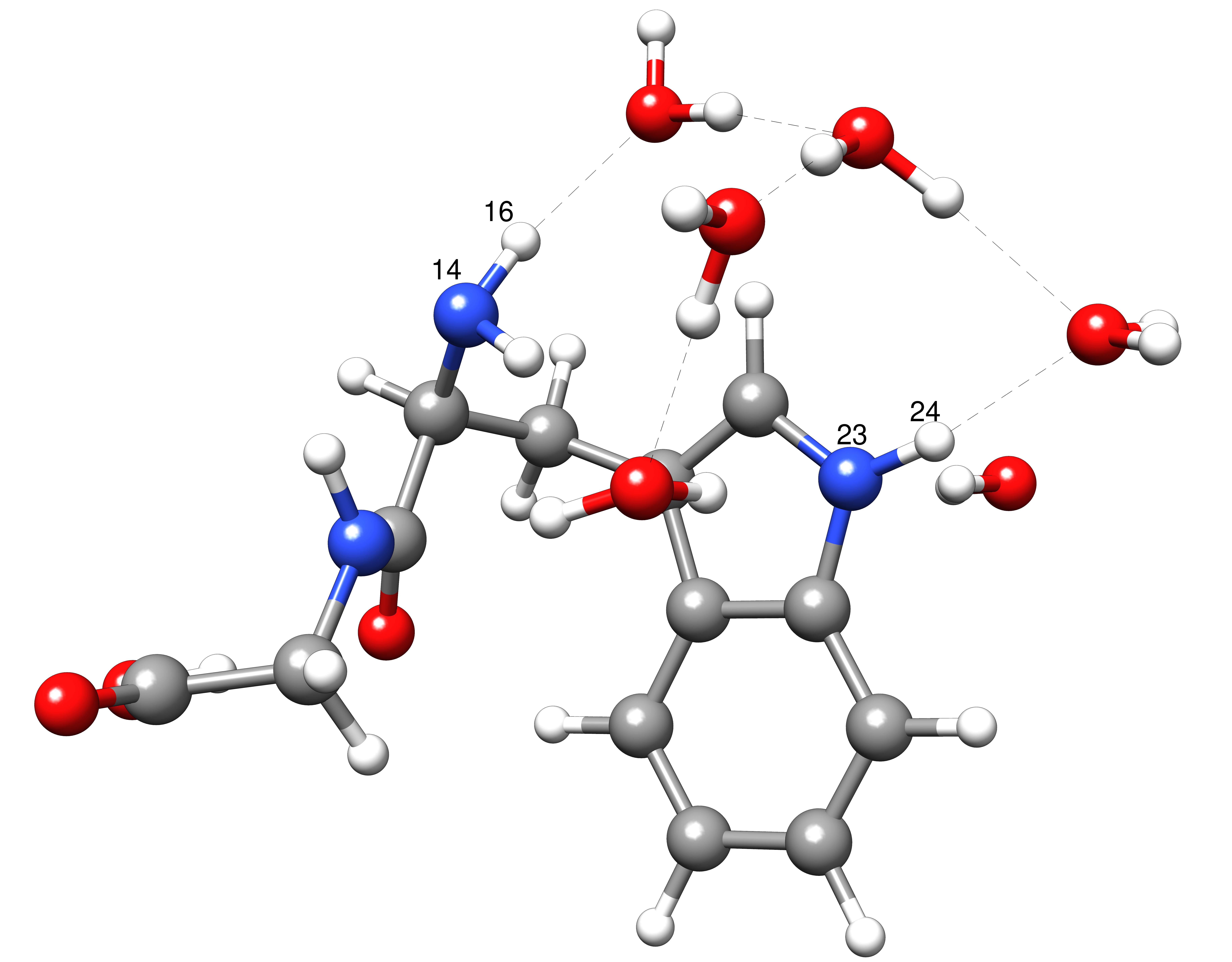
QCG Example 5
An example for solvating specific atoms and groups of the solute.
Solvating specific parts of the solute
Another feature of QCG is the solvation of user-defined positions of the solute. Therefore, the site-directed docking feature of the aISS algorithm available in xtb version 6.6.0 and above is used. of aISS is employed.
aISS algorithm is used instead of the xtbiff program. The aISS is invoked by default and generally recommended.To invoke the site-directed docking, the --directed <FILE> flag has to be used as a command line argument. In <FILE>, the list of solute atoms is given, at which solvent molecules should be added. Every line contains first the atom numbers of the solute followed by the number of solvent molecules that should be added at these positions (separated by a space bar or tab). If more solvent molecules are added than defined in this input, they are docked at overall energetically favored positions. This site-directed solvation can be used, for example, in microsolvation approaches or if we want to check the influence of a solvent molecule at different positions.
--wscal <real> keyword.As an example, a peptide is examined where only the NH group (23,24) of an aromatic ring should be investigated. To be safe, the nearby amine function (14,16) is also solvated and a short MD simulation of 10 ps is performed to sample the conformational space of solvents around this position.
crest struc.xyz --qcg water.xyz --nsolv 6 --gfnff --T 12 --alpb water --ensemble --md --mdtime 10 --wscal --enslvl gfnff 1.0
34
O 0.7908465 -0.4321715 -4.2643994
C -0.3831235 -0.9940515 -3.9568794
O -1.1148435 -1.4708015 -4.7910594
H 1.2267065 -0.1245415 -3.4325994
C -0.7173635 -1.0178815 -2.4672294
H 0.0087665 -1.6518615 -1.9533194
H -1.7049335 -1.4511115 -2.3552994
N -0.7078035 0.2896685 -1.8400494
H -1.5590335 0.7637185 -1.5684794
C 0.4192565 0.9435985 -1.5494294
O 1.5453865 0.5590385 -1.9069194
C 0.2526865 2.2075585 -0.7214394
H 0.7011065 3.0051485 -1.3150994
N -1.1580935 2.5068285 -0.4917194
H -1.4111935 2.2231185 0.4505406
H -1.3320535 3.4991085 -0.5697194
C 1.0738765 2.0542085 0.5798506
H 1.1502765 3.0377485 1.0469206
H 2.0800165 1.7306285 0.3130306
C 0.4326665 1.0962885 1.5265906
C -0.3971735 1.4372285 2.5787206
H -0.6540335 2.4144485 2.9561106
N -0.8729835 0.3020985 3.1869606
H -1.4698235 0.2838085 3.9950106
C -0.3649135 -0.8021715 2.5488706
C -0.5579935 -2.1606215 2.8096806
H -1.1968535 -2.4969215 3.6158506
C 0.1078865 -3.0652615 1.9975506
H -0.0163035 -4.1253915 2.1707006
C 0.9525165 -2.6269815 0.9583206
H 1.4700965 -3.3621015 0.3567306
C 1.1419165 -1.2791815 0.6973606
H 1.7986865 -0.9565015 -0.0988194
C 0.4658265 -0.3366915 1.4936606
23,24 5
14,16 1
--md keyword.The resulting structures contain the water molecules only around the functional group of interest even though this is not the overall energetically favored interaction site: