MECP Screening
(Semi-automated) Screening of benzene MECPs with GFN2-xTB
(Semi-automated) Screening of benzene MECPs with GFN2-xTB
An extension of the minimum energy crossing point optimization, is the metadynamics-based screening (analogously to conformational sampling). It can be called via the new input file reader . Again, for the GFN2-xTB S0/T1 MECPs of benzene:
crest --input input.toml
12
C 1.3830400000 -0.2213700000 0.0054100000
C 0.8812100000 1.0799600000 0.0137400000
C -0.4965300000 1.2961400000 0.0106300000
C -1.3728900000 0.2109800000 -0.0044700000
C -0.8710300000 -1.0904600000 -0.0146100000
C 0.5067700000 -1.3067000000 -0.0079300000
H 2.4566500000 -0.3899700000 0.0090900000
H 1.5639800000 1.9254500000 0.0228700000
H -0.8876100000 2.3099700000 0.0197800000
H -2.4463500000 0.3796100000 -0.0082500000
H -1.5536800000 -1.9359000000 -0.0272900000
H 0.8977800000 -2.3206600000 -0.0132700000
#This is a CREST input file
input = 'struc.xyz'
runtype='mecp_search' # MECP sampling runtype
#parallelization
threads = 10
#calculation data
[calculation]
eprint = true
elog="energies.log"
[[calculation.mecp]] # This block automatically sets up a uhf=0 and uhf=2 calculation with xtb
method = "xtb"
binary = "xtb"
flags = "--gfn 2 --grad"
[[calculation.constraints]]
gapdiff2 = [7.5, 0.005, 0.25]
#molecular dynamics data, required for MECP sampling runtype
[dynamics]
length = 10.0
tstep = 2.5
dump = 100.0
hmass = 4
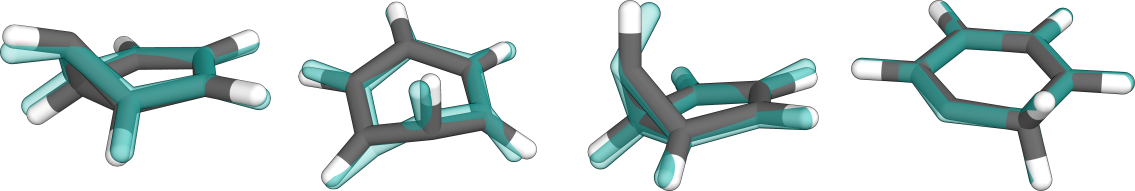
The sampling algorithm will run in parallel several (i.e., ten) metadynamics simulations on the mixed PES including the gap potential and MECP-optimize the output trajectories. While the metadynamics bias potentials are set up automatically, the user is responsible for setting the MD parameters, such as the simulation lengths in ps. Some benzene MECPs that can be found with this method are shown below (in comparison with FOMO-CASCI(6,5) structures in transparent blue).