Conformational Sampling
A simple conformational search.
Standard Conformational Search (iMTD-GC)
The default application of CREST is the iMTD-GC workflow. In the following, a standard production run with this workflow is shown for the alanineglycine molecule.
Let’s assume that we are interested in the conformations of Ala-Gly at the GFN2-xTB level with GBSA implicit solvation for water, and that we are using 4 CPU threads. Assuming further that our initial input structure (atomic coordinates, here in Ångström) are given in a file struc.xyz, then the conformational search can be initiated from the command line via:
crest struc.xyz --gfn2 --gbsa h2o -T 4--gfn2 specifies the usage of the SQM level GFN2-xTB, --gbsa h2o implements the GBSA implicit solvation for water, and -T 4 requests the usage of 4 CPU threads. You can save the terminal output of this command by adding > crest.out at the end of the line. The output will look something like the one in the output tab above.
20
C 2.081440 0.615100 -0.508430
C 2.742230 1.824030 -1.200820
N 4.117790 1.799870 -1.190410
C 4.943570 2.827040 -1.822060
C 6.440080 2.569360 -1.637600
O 7.351600 3.252270 -2.069090
N 0.610100 0.695090 -0.538780
O 2.095560 2.724940 -1.739670
O 6.705220 1.463410 -0.897460
H 0.303080 1.426060 0.103770
H 0.338420 1.050680 -1.460480
C 2.488753 -0.593400 -1.198448
H 2.416500 0.557400 0.532050
H 4.614100 1.081980 -0.670550
H 4.699850 3.794460 -1.373720
H 4.722890 2.844690 -2.894180
H 7.687400 1.448620 -0.860340
H 2.029201 -1.457008 -0.719999
H 2.170233 -0.542411 -2.238576
H 3.572730 -0.688405 -1.154998
==============================================
| |
| C R E S T |
| |
| Conformer-Rotamer Ensemble Sampling Tool |
| based on the GFN methods |
| P.Pracht, S.Grimme |
| Universitaet Bonn, MCTC |
==============================================
Version 2.11, Tue 13. Jul 16:11:14 CEST 2021
Using the xTB program. Compatible with xTB version 6.4.0
Cite work conducted with this code as
P. Pracht, F. Bohle, S. Grimme, PCCP, 2020, 22, 7169-7192.
and S. Grimme, JCTC, 2019, 15, 2847-2862.
with help from:
C.Bannwarth, F.Bohle, S.Ehlert, S.Grimme,
P.Pracht, S. Spicher
This program is distributed in the hope that it will be useful,
but WITHOUT ANY WARRANTY; without even the implied warranty of
MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE.
Command line input:
> crest struc.xyz --gfn2 --gbsa h2o -T 4
-gfn2 : Use of GFN2-xTB requested.
--gbsa h2o : implicit solvation
-T 4 (CPUs/Threads selected)
-------------------------
xTB Geometry Optimization
-------------------------
Geometry successfully optimized.
------------------------------------------------
Generating MTD length from a flexibility measure
------------------------------------------------
Calculating WBOs... done.
Calculating NCI flexibility... done.
covalent flexibility measure : 0.450
non-covalent flexibility measure : 0.823
flexibility measure : 0.501
t(MTD) / ps : 5.0
Σ(t(MTD)) / ps : 70.0 (14 MTDs)
-------------------------------------
Starting a trial MTD to test settings
-------------------------------------
Estimated runtime for one MTD (5.0 ps) on a single thread: 19 sec
Estimated runtime for a batch of 14 MTDs on 4 threads: 1 min 15 sec
list of Vbias parameters applied:
$metadyn 0.00300 1.300
$metadyn 0.00150 1.300
$metadyn 0.00075 1.300
$metadyn 0.00300 0.780
$metadyn 0.00150 0.780
$metadyn 0.00075 0.780
$metadyn 0.00300 0.468
$metadyn 0.00150 0.468
$metadyn 0.00075 0.468
$metadyn 0.00300 0.281
$metadyn 0.00150 0.281
$metadyn 0.00075 0.281
$metadyn 0.00100 0.100
$metadyn 0.00500 0.800
*******************************************************************************************
** N E W I T E R A T I O N C Y C L E **
*******************************************************************************************
========================================
MTD Iteration 1
========================================
========================================
| Meta-MD (MTD) Sampling |
========================================
<.......>
-----------------------
Multilevel Optimization
-----------------------
-------------------------
1. crude pre-optimization
-------------------------
Optimizing all 686 structures from file "crest_rotamers_0.xyz" ...
<.......>
done.
running RMSDs...
done.
E lowest : -33.87998
654 structures remain within 12.00 kcal/mol window
-------------------------------------
2. optimization with tight thresholds
-------------------------------------
Optimizing all 655 structures from file "crest_rotamers_1.xyz" ...
<.......>
done.
running RMSDs...
done.
E lowest : -33.88023
119 structures remain within 6.00 kcal/mol window
========================================
MTD Iteration 2
========================================
<.......>
<.......>
========================================
MTD Iterations done
========================================
Collecting ensmbles.
running RMSDs...
done.
E lowest : -33.88023
146 structures remain within 6.00 kcal/mol window
-----------------------------------------------
Additional regular MDs on lowest 4 conformer(s)
-----------------------------------------------
<.......>
Appending file crest_rotamers_1.xyz with new structures
-------------------------------------------
Ensemble optimization with tight thresholds
-------------------------------------------
Optimizing all 338 structures from file "crest_rotamers_1.xyz" ...
<.......>
done.
running RMSDs...
done.
E lowest : -33.88023
159 structures remain within 6.00 kcal/mol window
========================================
| Structure Crossing (GC) |
========================================
=============================
# threads = 4
=============================
input file name : crest_rotamers_3.xyz
number of atoms : 20
number of points on xyz files : 159
conformer energy window /kcal : 6.00
CN per atom difference cut-off : 0.3000
RMSD threshold : 0.2500
max. # of generated structures : 250
reading xyz file ...
# in E window 159
generating pairs ... 12719
66.0 % done
generated pairs : 10762
number of clash discarded : 1799
average rmsd w.r.t input : 2.82636
sd of ensemble : 0.63885
number of new structures : 98
removed identical structures : 402
<.......>
<.......>
================================================
| Final Geometry Optimization |
================================================
------------------------------------------------
Ensemble optimization with very tight thresholds
------------------------------------------------
Optimizing all 170 structures from file "crest_rotamers_4.xyz" ...
<.......>
done.
running RMSDs...
done.
E lowest : -33.88023
147 structures remain within 6.00 kcal/mol window
input file name : crest_rotamers_5.xyz
output file name : crest_rotamers_6.xyz
number of atoms : 20
number of points on xyz files : 170
RMSD threshold : 0.1250
Bconst threshold : 0.0100
population threshold : 0.0500
conformer energy window /kcal : 6.0000
# fragment in coord : 1
# bonds in reference structure : 19
number of reliable points : 170
reference state Etot : -33.8802280500000
number of doubles removed by rot/RMSD : 23
total number unique points considered further : 147
Erel/kcal Etot weight/tot conformer set degen origin
1 0.000 -33.88023 0.04207 0.25214 1 6 gc
2 0.000 -33.88023 0.04204 mtd1
3 0.001 -33.88023 0.04203 mtd9
4 0.001 -33.88023 0.04200 gc
5 0.001 -33.88023 0.04200 mtd3
6 0.001 -33.88023 0.04200 mtd9
7 0.050 -33.88015 0.03865 0.19312 2 5 mtd3
8 0.050 -33.88015 0.03864 mtd14
9 0.051 -33.88015 0.03862 mtd10
10 0.051 -33.88015 0.03861 md5
11 0.051 -33.88015 0.03860 mtd9
12 0.476 -33.87947 0.01884 0.09414 3 5 md6
13 0.476 -33.87947 0.01884 mtd12
14 ...
15 ...
<.......>
T /K : 298.15
E lowest : -33.88023
ensemble average energy (kcal) : 0.550
ensemble entropy (J/mol K, cal/mol K) : 34.054 8.139
ensemble free energy (kcal/mol) : -2.427
population of lowest in % : 25.214
number of unique conformers for further calc 61
list of relative energies saved as "crest.energies"
<.......>
CREST terminated normally.
xtb at the same level on which the conformational search shall be conducted. Since the input structure is taken as a reference for several topology checks within the sorting routine, such as unchanging coordination numbers of the atoms, providing a structure on the same level of theory is recommended.The program call first creates a coord file from the given input structure. Then, settings such as the length of the MTD simulation are automatically determined from a molecular flexibility measure. With these settings, the workflow as presented here is conducted.
The production run in this example yields 147 structures of Ala-Gly, distributed over 61 different conformers within 6 kcal/mol above the lowest conformer that was found at the search level. The three lowest conformers at the search level are shown below.
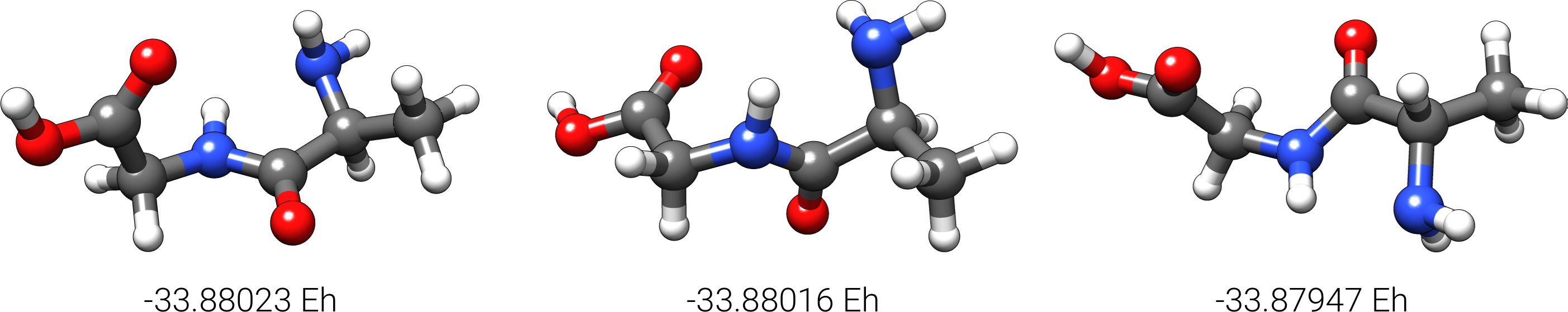
Each of the 61 conformers may shows several rotamers, which by definition are degenerate forms of the respective confromer. The final ensemble of all the found conformers is written to an ensemble file in the Xmol format called crest_conformers.xyz. The corresponding CRE, i.e., the ensemble containing conformers and rotamers is written to the file crest_rotamers.xyz.