MECP Optimization
Optimization of a benzene MECP with GFN2-xTB
CREST 3.0
Optimization of a benzene MECP with GFN2-xTB
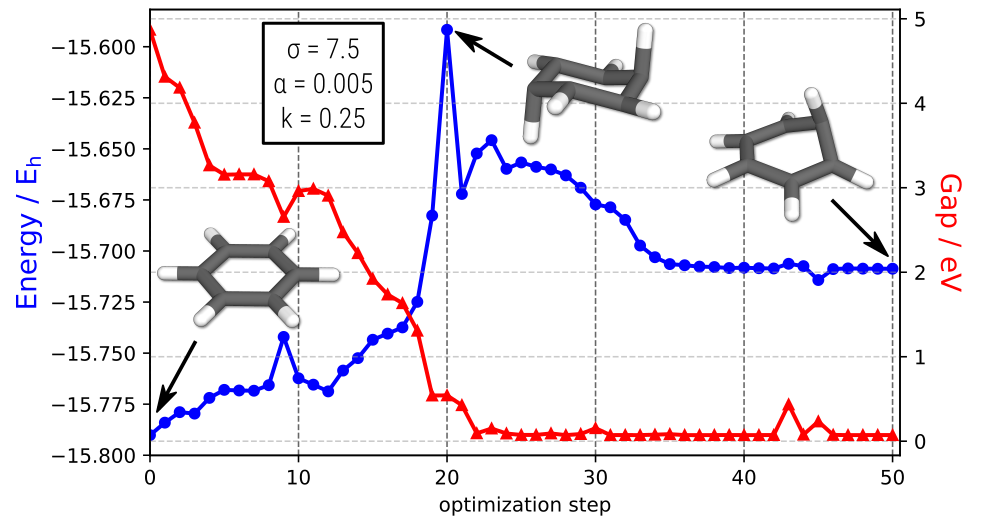
A specialized new feature of CREST 3 is the mixing of two potential energy surfaces with an additional bias potential to find minimum energy crossing points, as described in DOI: 10.1021/acs.jctc.2c00578. It can be called via the new input file reader , e.g., for the GFN2-xTB S0/T1 MECP of benzene:
crest --input input.toml
12
C 1.3830400000 -0.2213700000 0.0054100000
C 0.8812100000 1.0799600000 0.0137400000
C -0.4965300000 1.2961400000 0.0106300000
C -1.3728900000 0.2109800000 -0.0044700000
C -0.8710300000 -1.0904600000 -0.0146100000
C 0.5067700000 -1.3067000000 -0.0079300000
H 2.4566500000 -0.3899700000 0.0090900000
H 1.5639800000 1.9254500000 0.0228700000
H -0.8876100000 2.3099700000 0.0197800000
H -2.4463500000 0.3796100000 -0.0082500000
H -1.5536800000 -1.9359000000 -0.0272900000
H 0.8977800000 -2.3206600000 -0.0132700000
#This is a CREST input file
input = 'struc.xyz'
runtype='ancopt'
#parallelization
threads = 4
#calculation data
[calculation]
type = -1 # specify energy & gradient from [calculation.level] to be used
# -1 is for MECPs
eprint = true
elog="energies.log"
[[calculation.level]]
method = "xtb"
binary = "xtb"
uhf = 0 # GFN2-xTB S0 state
flags = "--gfn 2 --grad"
dir = 's0'
[[calculation.level]]
method = "xtb"
binary = "xtb"
uhf = 2 # GFN2-xTB T1 state
dir = 's1'
flags = "--gfn 2 --grad"
[[calculation.constraints]]
gapdiff2 = [10.0, 0.005, 0.25] #bias parameter to minimize the gap, [10, 0.005, 0.25] is ok for most molecules
The optimization trajectory (written to crestopt.log) will look like something in the following. Notice that the gap minimization between the two states (shown in red) is the initial driving force of the procedure.