Constrained Sampling
A guide to constrained conformational sampling.
Table of contents
--cinp command will generally be present for ALL xtb calculations; MD, MTD, optimizations, and singlepoints. Be careful not to bias your calculation too strongly and choose moderate force constants!Constrained conformational sampling
CREST’s conformational search relies on the quality of the underlying level of theory. Since we are choosing a SQM method (GFNn-xTB), it is possible for the system to freely form and break bonds. While it was shown in Example 2 how to handle the resulting topology mismatches in the sorting algorithm, in some occasions it might be required for the user to constrain certain parts of the geometry. In CREST this is possible by passing the respective constraints as a separate file to xtb.
A typical example are metal-organic compounds that can be sigificantly distorted at the GFNn-xTB level. Here, certain bond lengths or angles can be constrained in order to avoid this. An even simpler example are small non-covalent complexes. As was noted in Example 3, the MTD bias potential leads to dissociation of such non-covalently bound molecules. Instead of employing a wall potential like in the previous example, one can simply constrain some interatomic distances. This can be shown, for example for the methanol-acetamide complex from the S66 benchmark set.
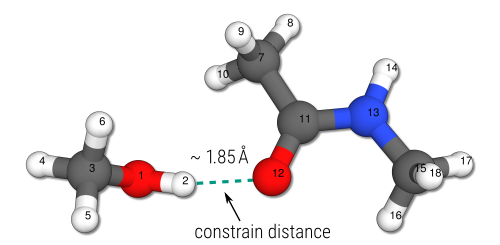
In the respective constraints.inp file, all constraints have to be specified in the xtb format. See the xtb Detailed Input documentation. For the methanol-acetamide example, the interatomic distance between the hyrdogen atom (2) and the oxygen atom (12) is constrained by an harmonic potential (force constant in atomic units, 0.25 Eh/Bohr2) to a value of 1.85 Ångström. The input files and the CREST command are given as
crest struc.xyz --cinp constraints.inp
18
O -2.3458343675 -0.5958980711 -1.0986703785
H -1.4259310467 -0.7659191523 -0.8402268081
C -3.1592303454 -1.1201985353 -0.0717185983
H -4.1945481644 -0.9177347321 -0.3348616664
H -3.0448003417 -2.2008537204 0.0422726914
H -2.9616880843 -0.6545221185 0.8983810538
C -0.1635664885 1.5241966295 0.3660367684
H 0.3428271404 2.4856457127 0.3311664119
H -0.5741106118 1.3780607546 1.3639134085
H -0.9932142903 1.5146833556 -0.3354687645
C 0.7520638942 0.3692578108 0.0501205567
O 0.3278691014 -0.7506529824 -0.2349609814
N 2.0782008097 0.6309820886 0.1093199208
H 2.3762132585 1.5414102214 0.4070071098
C 3.0606068433 -0.4091292185 -0.1134239002
H 2.7040496555 -1.0600665402 -0.9056008595
H 4.0009760815 0.0443946174 -0.4119376006
H 3.2201169562 -1.0136561201 0.7786516358
$constrain
force constant=0.25
distance: 2, 12, 1.85
$end
The respective conformational search provides an ensemble of non-covalently bound methanol-acetamide structures which all have a H(2)-O(12) distance close to 1.85 Å.
Fixing of entire substructure parts
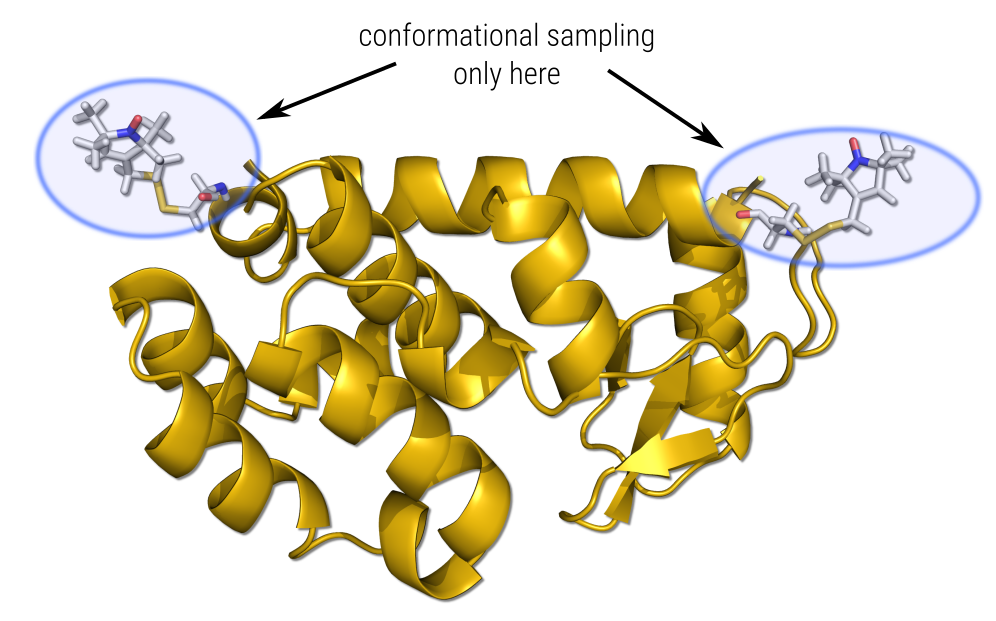
Sometimes it is necessary to fix entire parts of the structure. While complete freezing of atoms is not possible in CREST, putting a constraint on a large part of the substructure is possible. In principle, the procedure is identical to the one above , but needs some simple additions.
As an example, a fictional system consisting out of a linear n-octane chain with a diglycine substituent is calculated. Here, the entire n-octane chain shall be fixed, so that it remains linear.
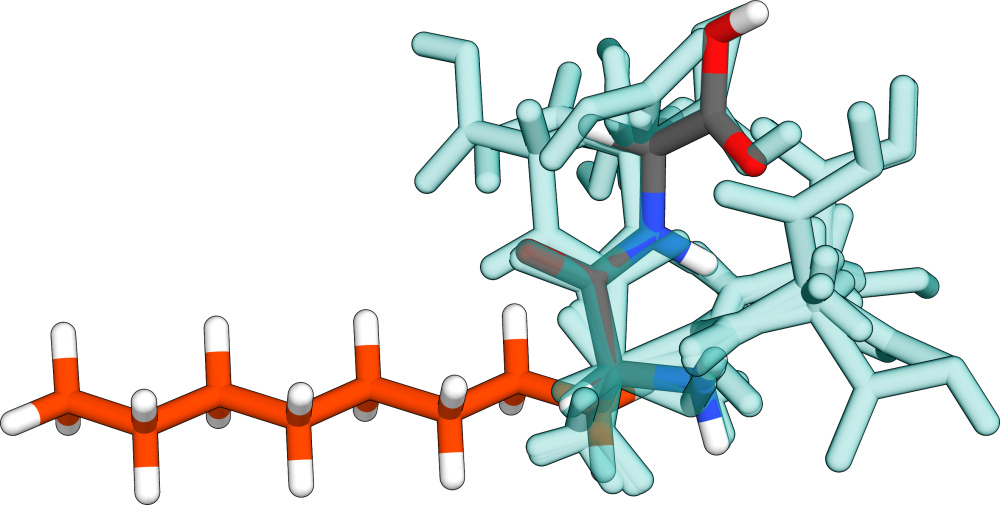
To prepare the calculation, several things have to be done:
- A constraints file has to be created
- In this file, all atoms that shall be fixed must be added to the
$constrainblock with theatoms:keyword - An unchanged reference geometry (= a copy of your input geometry) has to be added in the calculation directory and specified in the
$constrainblock with thereference=keyword - All atoms that are not constrained (= your side chain to be sampled) must be added to the
$metadynblock with theatoms:keyword - The command line argument
--subrmsdshould be used in the CREST call - (Optional) the MD/MTD time step should be reduced with
--tstep <REAL>
The final calculation and respective files will look like this:
crest fictional.xyz --cinp constraints.inp --subrmsd
41
C 1.0144800000 -0.0609700000 -0.0825900000
C 2.5333400000 -0.0608400000 -0.0710600000
C 3.0664700000 -0.3643600000 1.3256100000
C 4.5908500000 -0.3677000000 1.3420700000
C 5.1165500000 -0.6739300000 2.7411100000
C 6.6413400000 -0.6756000000 2.7574300000
C 7.1736700000 -0.9813000000 4.1534600000
C 8.6899400000 -0.9817400000 4.1630900000
H 0.6423500000 0.1573500000 -1.0871300000
H 0.6205000000 0.6974700000 0.6016700000
H 0.6236000000 -1.0367300000 0.2236500000
H 2.9003600000 0.9164900000 -0.4063100000
H 2.9034800000 -0.8084900000 -0.7825300000
H 2.6926100000 -1.3400300000 1.6597300000
H 2.6921400000 0.3848400000 2.0340900000
H 4.9664500000 0.6088800000 1.0113000000
H 4.9674500000 -1.1157300000 0.6334000000
H 4.7415200000 -1.6506000000 3.0706100000
H 4.7391300000 0.0736300000 3.4494600000
H 7.0139700000 0.3012200000 2.4251500000
H 7.0160900000 -1.4228100000 2.0470400000
H 6.8073200000 -1.9587400000 4.4877200000
H 6.8053100000 -0.2343600000 4.8660200000
C 9.2154250000 -1.9852880000 3.2579620000
H 9.0652900000 -1.2016200000 5.1680800000
H 9.0847000000 -0.0064100000 3.8597300000
N 10.6547150000 -1.9569240000 3.2931620000
C 8.7477200000 -3.2959450000 3.6653100000
H 8.8728820000 -1.7760900000 2.2456280000
H 11.0200180000 -2.6545630000 2.6639420000
H 10.9798510000 -1.0457920000 3.0099850000
O 9.0775760000 -4.2852730000 3.0322020000
N 7.8939040000 -3.4298300000 4.8171220000
H 7.7233110000 -2.5201690000 5.2164720000
C 6.6427270000 -4.0223990000 4.4208670000
C 5.7829810000 -4.1572140000 5.5806780000
H 6.1579230000 -3.3865210000 3.6815810000
H 6.8270280000 -5.0051580000 3.9894270000
O 4.5047420000 -4.7116320000 5.4438410000
O 6.1735030000 -3.7875090000 6.6757630000
H 4.0745330000 -4.7284110000 6.2906810000
$constrain
atoms: 1-26
force constant=0.5
reference=coord.ref
$metadyn
atoms: 27-41
$end
==============================================
| |
| C R E S T |
| |
| Conformer-Rotamer Ensemble Sampling Tool |
| based on the GFN methods |
| P.Pracht, S.Grimme |
| Universitaet Bonn, MCTC |
==============================================
Version 2.12, Thu 19. Mai 16:32:32 CEST 2022
Using the xTB program. Compatible with xTB version 6.4.0
Cite work conducted with this code as
• P.Pracht, F.Bohle, S.Grimme, PCCP, 2020, 22, 7169-7192.
• S.Grimme, JCTC, 2019, 15, 2847-2862.
and for works involving QCG as
• S.Spicher, C.Plett, P.Pracht, A.Hansen, S.Grimme,
JCTC, 2022, 18 (5), 3174-3189.
with help from:
C.Bannwarth, F.Bohle, S.Ehlert, S.Grimme,
C.Plett, P.Pracht, S.Spicher
This program is distributed in the hope that it will be useful,
but WITHOUT ANY WARRANTY; without even the implied warranty of
MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE.
Command line input:
> crest fictional.xyz --cinp constraints.inp --subrmsd
-cinp : constraints.inp
'constraints.inp' file present.
content of the constraining file (sorted):
> $constrain
> atoms: 1-26
> force constant=0.5
> reference=coord.ref
> $metadyn
> atoms: 27-41
fix file: coord.ref
atoms: 27-41
# of atoms considered for RMSDs:15
-------------------------
xTB Geometry Optimization
-------------------------
Geometry successfully optimized.
------------------------------------------------
Generating MTD length from a flexibility measure
------------------------------------------------
Calculating WBOs... done.
Calculating NCI flexibility... done.
covalent flexibility measure : 0.555
non-covalent flexibility measure : 0.778
flexibility measure : 0.567
t(MTD) / ps : 5.0
Σ(t(MTD)) / ps : 70.0 (14 MTDs)
-------------------------------------
Starting a trial MTD to test settings
-------------------------------------
Trial MTD 1 did not converge!
Reducing the time step to 4 fs and trying again...
Trial MTD 2 did not converge!
Reducing the time step to 3 fs and trying again...
Trial MTD 3 did not converge!
Reducing the time step to 2 fs and trying again...
Estimated runtime for one MTD (5.0 ps) on a single thread: 50 sec
Estimated runtime for a batch of 14 MTDs on 4 threads: 3 min 21 sec
list of Vbias parameters applied:
$metadyn 0.00300 1.300
$metadyn 0.00150 1.300
$metadyn 0.00075 1.300
$metadyn 0.00300 0.780
$metadyn 0.00150 0.780
$metadyn 0.00075 0.780
$metadyn 0.00300 0.468
$metadyn 0.00150 0.468
$metadyn 0.00075 0.468
$metadyn 0.00300 0.281
$metadyn 0.00150 0.281
$metadyn 0.00075 0.281
$metadyn 0.00100 0.100
$metadyn 0.00500 0.800
*******************************************************************************************
** N E W I T E R A T I O N C Y C L E **
*******************************************************************************************
[....]
[....]
input file name : crest_rotamers_6.xyz
output file name : crest_rotamers_7.xyz
number of atoms : 41
atoms included in RMSD : 15
number of points on xyz files : 148
RMSD threshold : 0.1250
Bconst threshold : 0.0100
population threshold : 0.0500
conformer energy window /kcal : 6.0000
# fragment in coord : 1
# bonds in reference structure : 40
number of reliable points : 148
reference state Etot : -56.0076698600000
number of doubles removed by rot/RMSD : 8
total number unique points considered further : 140
Erel/kcal Etot weight/tot conformer set degen origin
1 0.000 -56.00767 0.07792 0.15581 1 2 md2
2 0.000 -56.00767 0.07789 mtd10
3 0.139 -56.00745 0.06163 0.12319 2 2 md4
4 0.140 -56.00745 0.06157 mtd10
5 0.289 -56.00721 0.04784 0.04784 3 1 mtd10
6 0.339 -56.00713 0.04398 0.08793 4 2 mtd10
7 0.340 -56.00713 0.04395 md8
8 0.371 -56.00708 0.04170 0.04170 5 1 mtd9
9 0.414 -56.00701 0.03875 0.03875 6 1 hor
10 0.705 -56.00655 0.02373 0.02373 7 1 hor
11 0.764 -56.00645 0.02147 0.02147 8 1 hor
12 0.771 -56.00644 0.02124 0.02124 9 1 mtd2
13 0.797 -56.00640 0.02031 0.02031 10 1 mtd4
14 0.801 -56.00639 0.02017 0.02017 11 1 mtd10
15 0.915 -56.00621 0.01665 0.01665 12 1 mtd10
[....]
138 5.912 -55.99825 0.00000 gc
139 5.928 -55.99822 0.00000 0.00000 120 1 hor
140 5.994 -55.99812 0.00000 0.00000 121 1 gc
T /K : 298.15
E lowest : -56.00767
ensemble average energy (kcal) : 0.744
ensemble entropy (J/mol K, cal/mol K) : 31.656 7.566
ensemble free energy (kcal/mol) : -2.256
population of lowest in % : 15.581
number of unique conformers for further calc 121
list of relative energies saved as "crest.energies"
-----------------
Wall Time Summary
-----------------
test MD wall time : 0h : 0m :15s
MTD wall time : 0h : 4m :18s
multilevel OPT wall time : 0h : 5m :28s
MD wall time : 0h : 1m :14s
GC wall time : 0h : 1m : 0s
--------------------
Overall wall time : 0h :12m :24s
A large ensemble of 121 unique conformers was obtained for our fictional system. Inspection of these structures reveals that, indeed, all of them still show a linear conformation of the n-octane chain.
Semi-automated preparation of a constraint input
The constraint.inp file from the previous section can actually be prepared by CREST. The respective command is
crest fictional.xyz --constrain 1-26
Here, the --constrain <atomlist> command was used, which will simply write a file called .xcontrol.sample (=constraints.inp from above). The command will automatically make a copy of the input geometry and name it coord.ref. All atoms not present in <atomlist> will be added to the metadynamics bias.
Automated bond constraints
CREST also has a function for automatically constraining the interatomic distances of all covalent bonds to those from the input structure. This option can be invoked with the --cbonds command (or its variants, see Keyword Documentation ).
crest struc.xyz --cbonds
The only drawback here is, that the information whether an interatomic distance corresponds to a covalent bond is approximated from an empirical topology set up from atomic coordination numbers. It can not be ensured that the constrained distances actually correspond to the “true” covalent bonds. Furthermore, metal atoms are often problematic due to their large variety of coordination numbers.