File Formats
Examples for input files and formats.
Table of contents
- Input Atomic Coordinates
- Ensemble and Trajectory Files
- Atomlists
- Constraints
- Vibrational frequencies in the
vibspectrumformat
Input Atomic Coordinates
The program supports molecular [INPUT] files in the
- Turbomole coord format (.coord extension, Bohr)
- Xmol format (.xyz extension, Ångström)
- MDL molfile format (V2000,V3000, .sdf/.mol extension, Ångström)
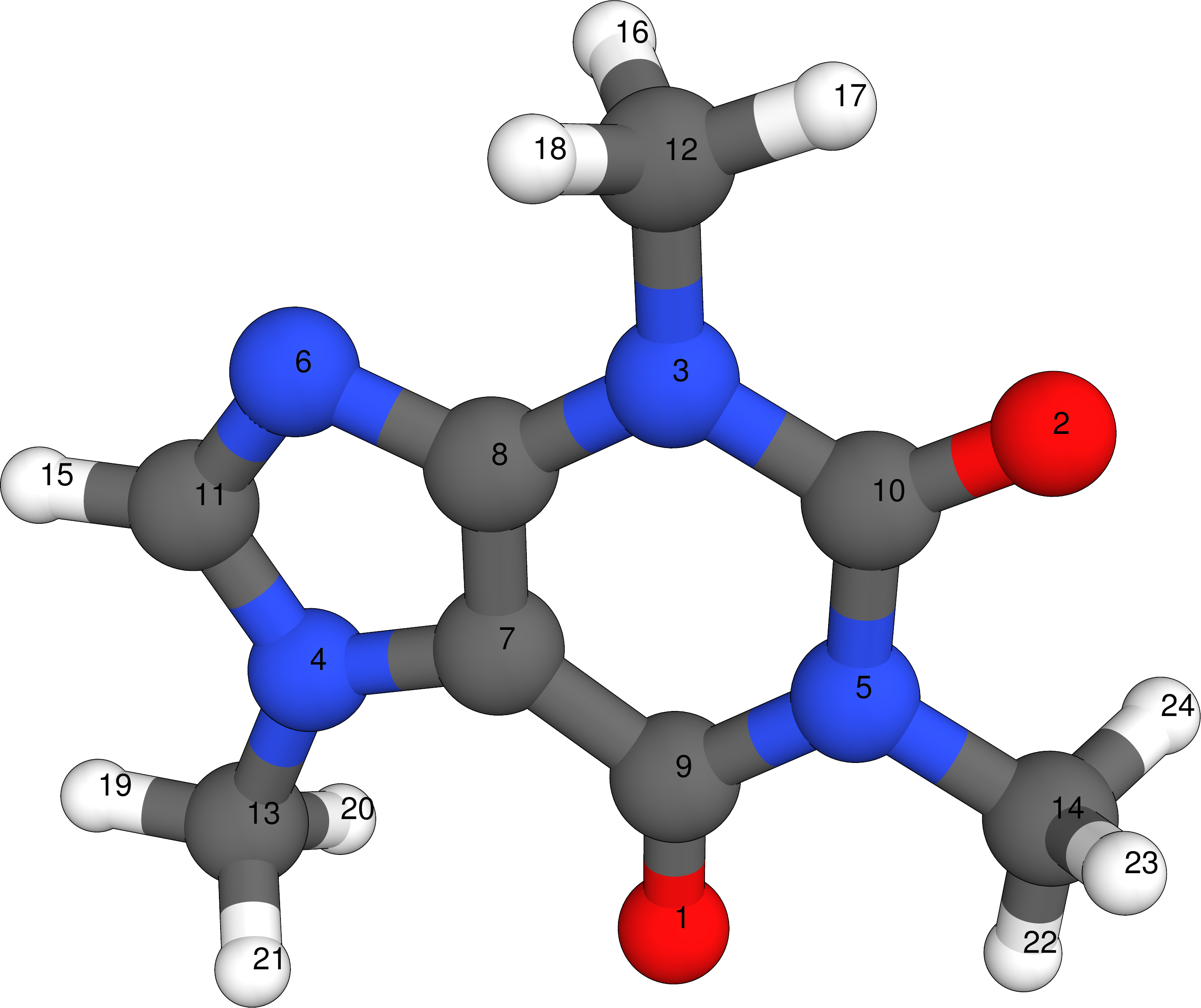
Example coordinates are shown for the caffeine molecule below. Atoms are typically specified line-by-line, each containing the Cartesian x-, y-, and z-coordinate and the atom type by its element symbol. Depending on the input file type the Cartesian coordinates can be in atomic units (Bohr) or Ångström. Some file formats additionally require the total number of atoms in the molecule. The order in which atoms are specified should not be changed since structure comparisons depend on it. For the caffeine example the atom order has been marked in the below figure for better comprehension.
$coord
0.88817153184985 4.85432985322531 0.00113383599811 O
-5.90936424946312 -0.83828274793318 -0.00056691799905 O
-1.83038924627610 -2.48026624585729 0.00000000000000 N
4.19179168499859 0.26682940488766 -0.00056691799905 N
-2.54678462441286 2.04033787859209 -0.00018897266635 N
2.66810507621022 -3.66077849255219 0.00037794533270 N
1.62119650462550 0.48981715118187 -0.00151178133081 C
0.73642648076997 -1.93961544742699 -0.00075589066540 C
0.05801460856977 2.68719131551167 -0.00113383599811 C
-3.60200799331702 -0.47148680254582 -0.00075589066540 C
4.73036378409903 -2.26729405087968 0.00056691799905 C
-2.69777378482733 -5.09470308482382 0.00151178133081 C
6.03314134592305 2.27919932885980 0.00056691799905 C
-4.34051317341685 4.13491091242693 0.00132280866446 C
6.64484586690134 -2.98331148368374 0.00151178133081 H
-1.97495333603463 -6.04202306124154 -1.68884871917917 H
-4.75946557471709 -5.21488970062308 0.00207869932986 H
-1.97419744536923 -6.04013333457803 1.69262817250619 H
7.93534020541253 1.47417577020440 0.00037794533270 H
5.75761919838324 3.41889347962287 -1.69924221582848 H
5.75724125305054 3.41719272562571 1.70150988782469 H
-3.41794861629111 5.98117386267651 -0.00056691799905 H
-5.54105652274496 3.97352825536315 1.67826624986351 H
-5.54559186673738 3.97239441936504 -1.67221912454028 H
$end
24
(in this line a comment or an energy can be placed)
O 0.470000 2.568800 0.000600
O -3.127100 -0.443600 -0.000300
N -0.968600 -1.312500 0.000000
N 2.218200 0.141200 -0.000300
N -1.347700 1.079700 -0.000100
N 1.411900 -1.937200 0.000200
C 0.857900 0.259200 -0.000800
C 0.389700 -1.026400 -0.000400
C 0.030700 1.422000 -0.000600
C -1.906100 -0.249500 -0.000400
C 2.503200 -1.199800 0.000300
C -1.427600 -2.696000 0.000800
C 3.192600 1.206100 0.000300
C -2.296900 2.188100 0.000700
H 3.516300 -1.578700 0.000800
H -1.045100 -3.197300 -0.893700
H -2.518600 -2.759600 0.001100
H -1.044700 -3.196300 0.895700
H 4.199200 0.780100 0.000200
H 3.046800 1.809200 -0.899200
H 3.046600 1.808300 0.900400
H -1.808700 3.165100 -0.000300
H -2.932200 2.102700 0.888100
H -2.934600 2.102100 -0.884900
2519
caffeine (this is a comment line)
24 25 0 0 0 0 0 0 0999 V2000
0.4700 2.5688 0.0006 O 0 0 0 0 0 0 0 0 0 0 0 0
-3.1271 -0.4436 -0.0003 O 0 0 0 0 0 0 0 0 0 0 0 0
-0.9686 -1.3125 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
2.2182 0.1412 -0.0003 N 0 0 0 0 0 0 0 0 0 0 0 0
-1.3477 1.0797 -0.0001 N 0 0 0 0 0 0 0 0 0 0 0 0
1.4119 -1.9372 0.0002 N 0 0 0 0 0 0 0 0 0 0 0 0
0.8579 0.2592 -0.0008 C 0 0 0 0 0 0 0 0 0 0 0 0
0.3897 -1.0264 -0.0004 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0307 1.4220 -0.0006 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.9061 -0.2495 -0.0004 C 0 0 0 0 0 0 0 0 0 0 0 0
2.5032 -1.1998 0.0003 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.4276 -2.6960 0.0008 C 0 0 0 0 0 0 0 0 0 0 0 0
3.1926 1.2061 0.0003 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.2969 2.1881 0.0007 C 0 0 0 0 0 0 0 0 0 0 0 0
3.5163 -1.5787 0.0008 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.0451 -3.1973 -0.8937 H 0 0 0 0 0 0 0 0 0 0 0 0
-2.5186 -2.7596 0.0011 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.0447 -3.1963 0.8957 H 0 0 0 0 0 0 0 0 0 0 0 0
4.1992 0.7801 0.0002 H 0 0 0 0 0 0 0 0 0 0 0 0
3.0468 1.8092 -0.8992 H 0 0 0 0 0 0 0 0 0 0 0 0
3.0466 1.8083 0.9004 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.8087 3.1651 -0.0003 H 0 0 0 0 0 0 0 0 0 0 0 0
-2.9322 2.1027 0.8881 H 0 0 0 0 0 0 0 0 0 0 0 0
-2.9346 2.1021 -0.8849 H 0 0 0 0 0 0 0 0 0 0 0 0
1 9 2 0 0 0 0
2 10 2 0 0 0 0
3 8 1 0 0 0 0
3 10 1 0 0 0 0
3 12 1 0 0 0 0
4 7 1 0 0 0 0
4 11 1 0 0 0 0
4 13 1 0 0 0 0
5 9 1 0 0 0 0
5 10 1 0 0 0 0
5 14 1 0 0 0 0
6 8 1 0 0 0 0
6 11 2 0 0 0 0
7 8 2 0 0 0 0
7 9 1 0 0 0 0
11 15 1 0 0 0 0
12 16 1 0 0 0 0
12 17 1 0 0 0 0
12 18 1 0 0 0 0
13 19 1 0 0 0 0
13 20 1 0 0 0 0
13 21 1 0 0 0 0
14 22 1 0 0 0 0
14 23 1 0 0 0 0
14 24 1 0 0 0 0
M END
$$$$
Ensemble and Trajectory Files
Ensemble (.xyz) and trajectory (.trj) files in CREST are given in the .xyz format as specified above. The files simply consists out of all the structures pasted after another.
If the ensemble is to be processed by CREST, e.g., with the CREGEN sorting routine, an energy in atomic units (Eh, Hartree) should be provided in the comment line. An example for an n-butane ensemble can be seen here:
14
-1.95933513
C -1.9450668421 0.1311167672 -0.0001772545
C -0.5674743975 -0.5132672899 0.0001933101
C 0.5674752787 0.5132638115 0.0001915171
C 1.9450638093 -0.1311175361 -0.0001779077
H -2.0853782627 0.7646639932 0.8782051254
H -2.7466842224 -0.6116573212 -0.0004139053
H -2.0848832626 0.7646834243 -0.8786240987
H -0.4586079390 -1.1740559621 -0.8706321703
H -0.4589068044 -1.1737556210 0.8712831745
H 0.4588981405 1.1737728909 0.8712658721
H 0.4586137126 1.1740687810 -0.8706234929
H 2.0850227749 -0.7644118141 -0.8787988057
H 2.0852599026 -0.7649044717 0.8780496213
H 2.7466915989 0.6116467123 -0.0000648118
14
-1.95861370
C -1.5634429064 0.0307829908 0.5671856584
C -0.6780400299 -0.3575310612 -0.6072767893
C 0.6780374832 0.3575328640 -0.6072807936
C 1.5634444854 -0.0307786855 0.5671868578
H -1.7381548026 1.1085042885 0.5904194743
H -1.1085086806 -0.2426226956 1.5210355320
H -2.5399702240 -0.4581336702 0.5235479812
H -1.2059135035 -0.1493891533 -1.5491742664
H -0.5130824651 -1.4435518231 -0.6080674515
H 0.5130888273 1.4435548434 -0.6080686466
H 1.2059008586 0.1493974488 -1.5491848268
H 1.1087081819 0.2429933207 1.5210259608
H 2.5401620514 0.4577301888 0.5232639016
H 1.7377812886 -1.1085555327 0.5906966592
14
-1.95476831
C -1.8280514558 0.1026283679 -0.3378109356
C -0.6096777513 -0.4807999039 0.3626338277
C 0.6096679585 0.4807861256 0.3626336492
C 1.8280554740 -0.1026120684 -0.3378127654
H -2.6737183814 -0.5895996214 -0.3384876099
H -1.6051787878 0.3482893729 -1.3783393521
H -2.1588537968 1.0236078559 0.1469845806
H -0.3239394543 -1.4268049722 -0.1182711212
H -0.8704029060 -0.7659987748 1.3922403082
H 0.8703773548 0.7659540027 1.3922522276
H 0.3239374558 1.4268002601 -0.1182574316
H 1.6052556663 -0.3480468876 -1.3784082402
H 2.1587507058 -1.0237028671 0.1468484919
H 2.6738409543 0.5894715887 -0.3381226983
Atomlists
Atomlists are used for some argument such as --notopo. They are used to specify atoms or types of atoms of your input structure. Effectively, atomlists are a single string consisting out of the respective number of the atom, or the element symbol. The only important thing to notice is that no whitespaces should be present in the atomlist, and arguments are separated by a comma ,. As an example,
1,3,4,5,6
would select atom 1 (one of the oxygens) and all the nitrogen atoms in the caffeine exmple from above. Alternatively a range of atoms could be specified, as for example
1,3-6
Finally, atomlists are compatible with element symbols, so the same atoms would be selected by
1,n
Constraints
Files for constraining must be in xtb’s input format. An example would be
$constrain
atoms: 1-26
force constant=0.5
reference=coord.ref
$metadyn
atoms: 27-41
$end
See Example 4 for a more detailed guide.
Vibrational frequencies in the vibspectrum format
Construction of vibspectrum files is straightforward. They are declared by the $vibrational spectrum keyword and ended by the $end keyword. Within this block, each mode is assigned one line containing the mode number, symmetry label, frequency in cm-1, intensity (arbitrary units), in that order. For a molecule with N atoms, 3N lines have to be present. The modes must be arragend to their frequencies in ascending order. Comment lines starting with # are ignored.
For thymine (15 atoms), a vibspectrum file would look something like this:
$vibrational spectrum
# mode symmetry wave number IR intensity selection rules
# cm**(-1) (km*mol⁻¹) IR RAMAN
1 -0.00 0.00000 - -
2 -0.00 0.00000 - -
3 -0.00 0.00000 - -
4 0.00 0.00000 - -
5 0.00 0.00000 - -
6 0.00 0.00000 - -
7 a 70.05 0.38846 YES YES
8 a 105.13 0.44816 YES YES
9 a 147.88 4.81587 YES YES
10 a 261.06 3.14819 YES YES
11 a 301.90 0.50209 YES YES
12 a 360.79 7.68669 YES YES
13 a 377.25 25.99351 YES YES
14 a 431.27 13.50432 YES YES
15 a 522.33 2.07831 YES YES
16 a 583.19 4.66771 YES YES
17 a 587.91 102.69438 YES YES
18 a 664.09 13.87677 YES YES
19 a 685.26 10.36450 YES YES
20 a 720.79 217.35379 YES YES
21 a 755.25 2.31369 YES YES
22 a 777.49 2.22428 YES YES
23 a 860.72 15.31103 YES YES
24 a 958.79 1.79962 YES YES
25 a 1006.53 1.51584 YES YES
26 a 1035.98 6.36000 YES YES
27 a 1126.59 38.23933 YES YES
28 a 1153.10 45.04718 YES YES
29 a 1212.04 55.76099 YES YES
30 a 1273.30 29.07111 YES YES
31 a 1303.81 96.17203 YES YES
32 a 1327.48 68.41479 YES YES
33 a 1400.54 60.93027 YES YES
34 a 1410.68 139.45727 YES YES
35 a 1454.52 5.40728 YES YES
36 a 1472.57 1.05534 YES YES
37 a 1663.21 141.68596 YES YES
38 a 1739.67 685.35070 YES YES
39 a 1779.98 830.38911 YES YES
40 a 3016.25 8.53182 YES YES
41 a 3034.18 7.87679 YES YES
42 a 3052.46 20.26137 YES YES
43 a 3076.43 17.81182 YES YES
44 a 3398.16 26.97265 YES YES
45 a 3444.01 49.06620 YES YES
$end
Note that some additional information was present (IR and RAMAN coloumns), which is ignored by CREST. Not also, that the first six entries correspond to the translation and rotation and hence have a frequency of zero.